How to Cite SC-MO-GRN-DB
If you use SC-MO-GRN-DB in your research, please cite:
Valensi H, Karamveer K, Moeller E, Ozdogan SE, Edwards RM, Uzun Y. SC-MO-GRN-DB: A comprehensive repository for single-cell multiomic gene regulatory networks. iScience. 2026;29(4):115323. doi:10.1016/j.isci.2026.115323
Data Content
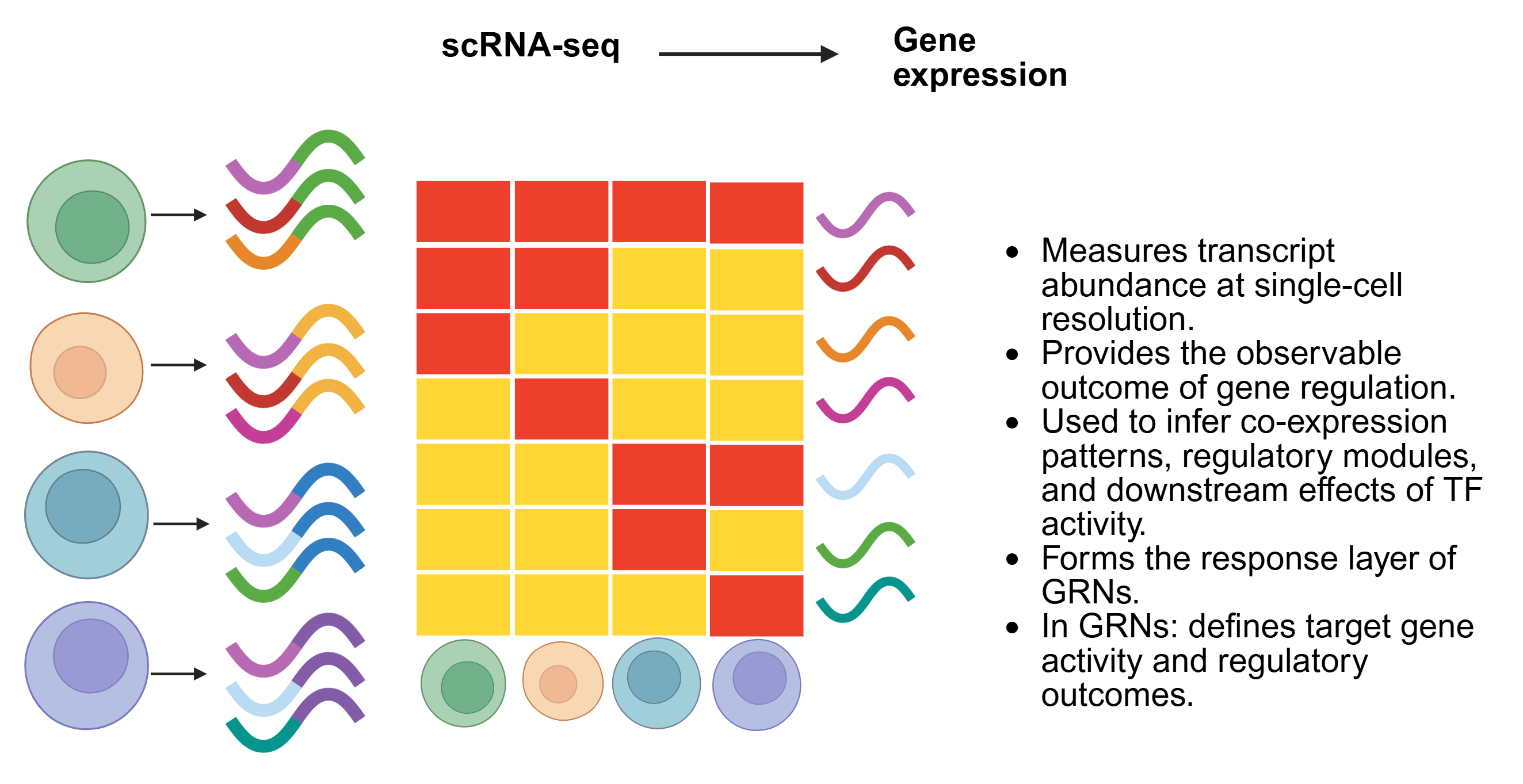
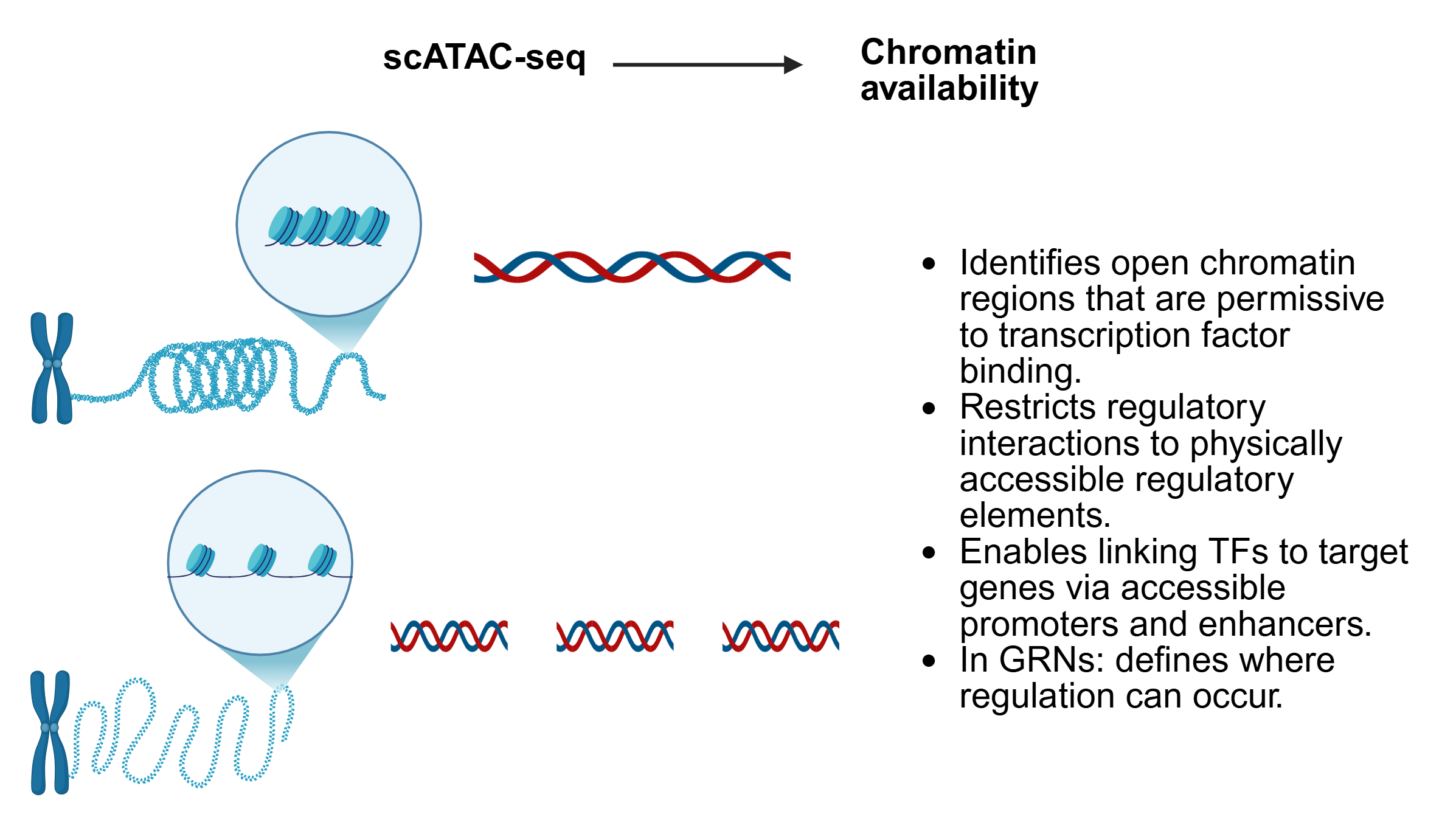
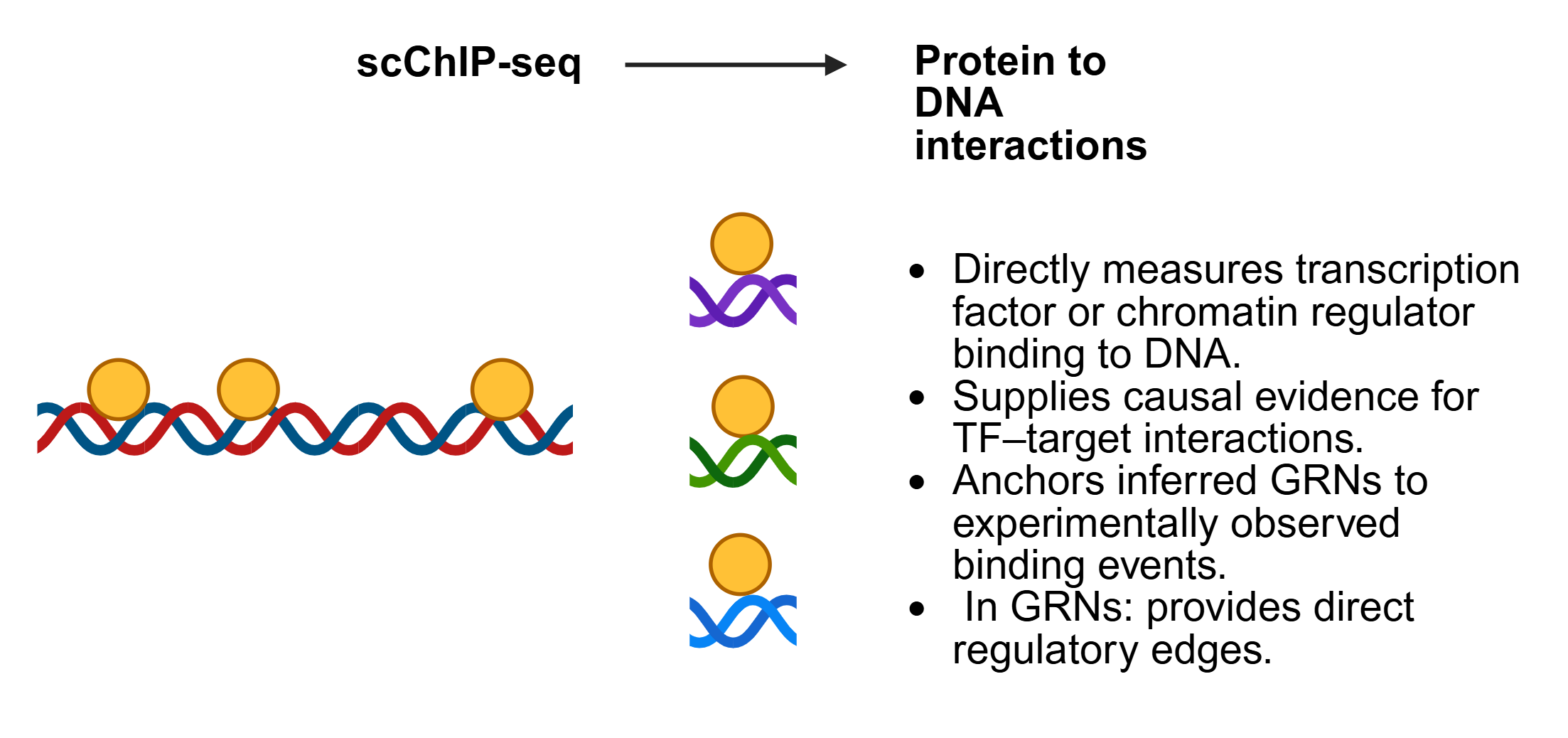
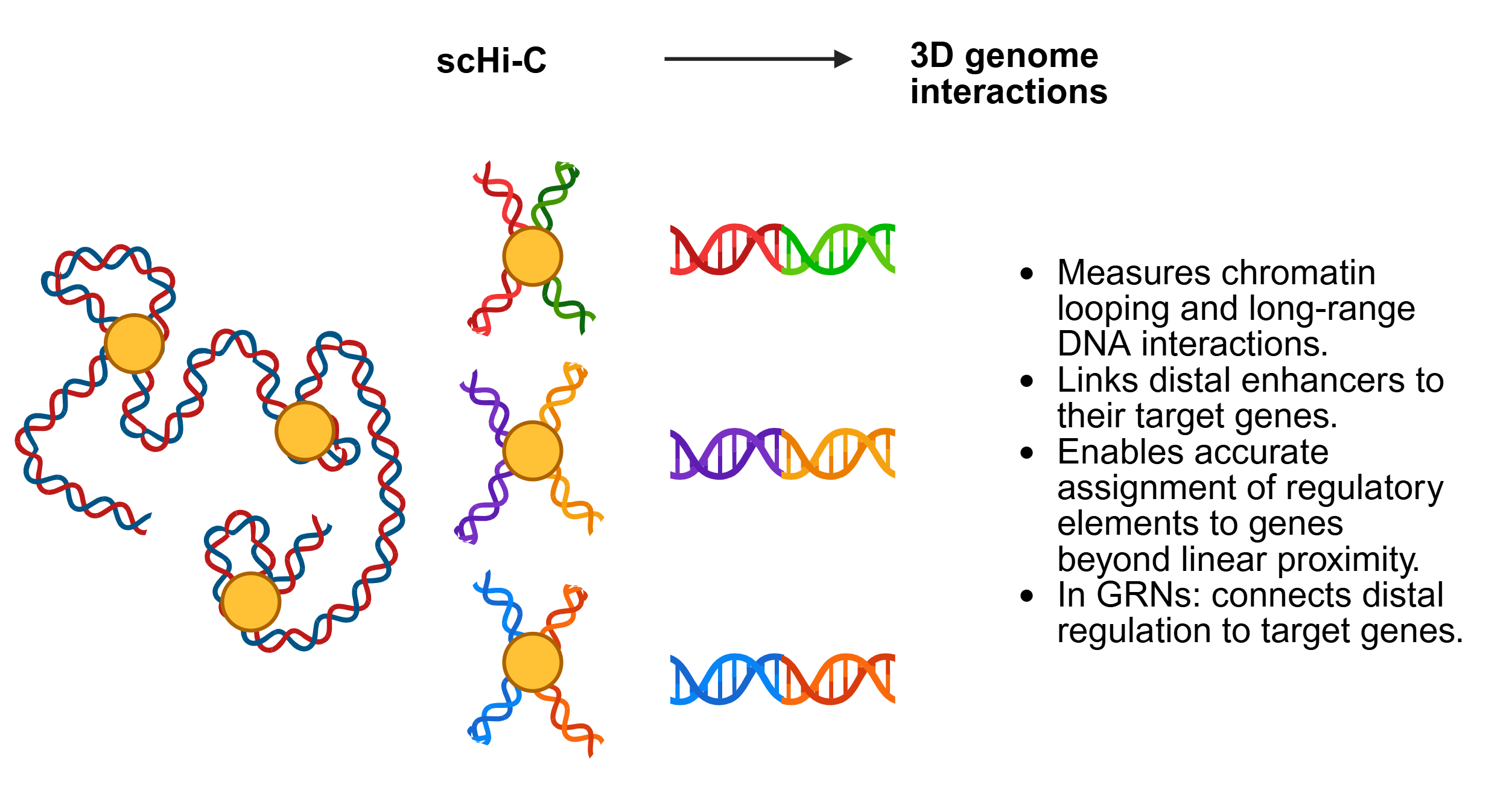
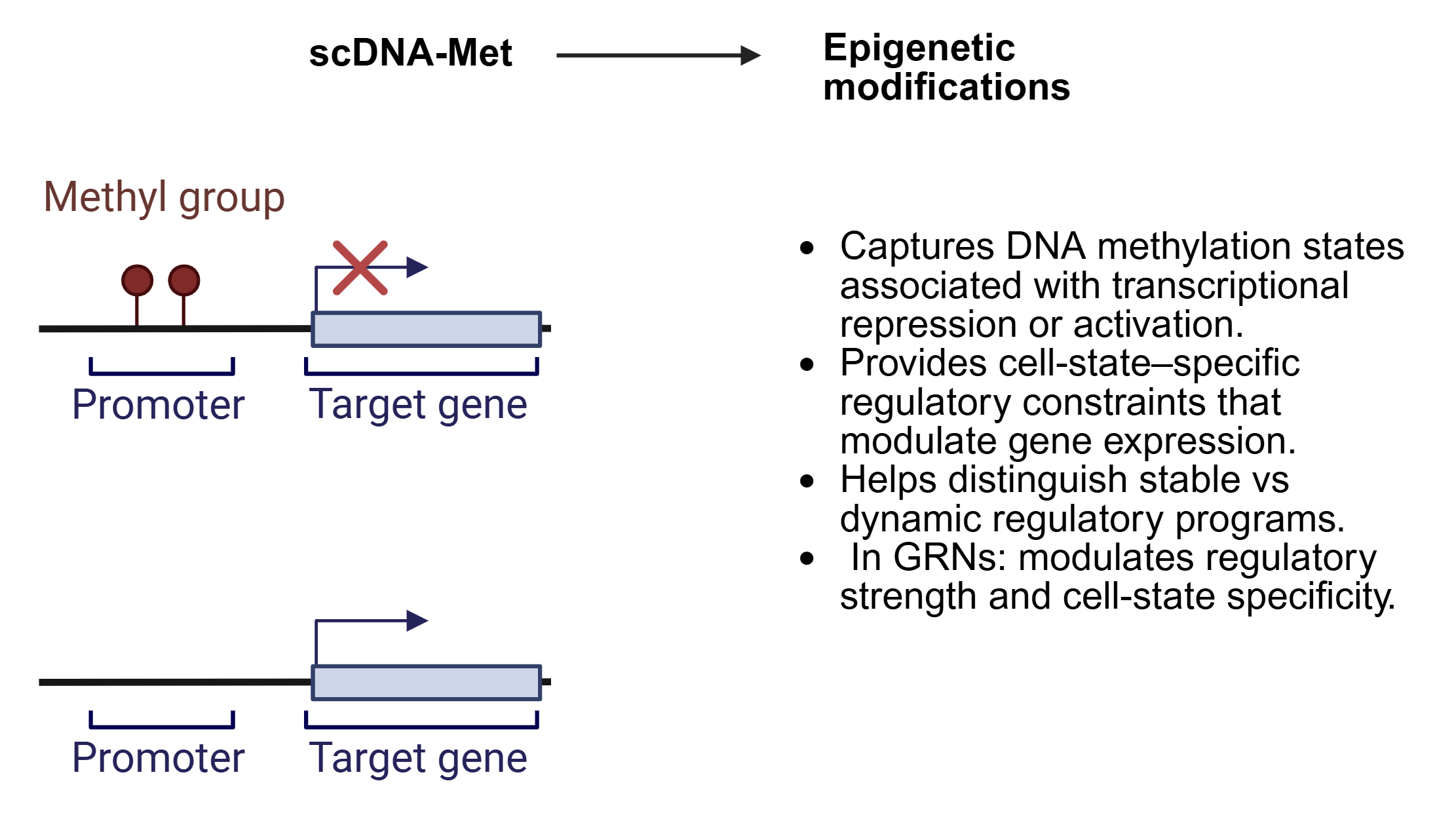
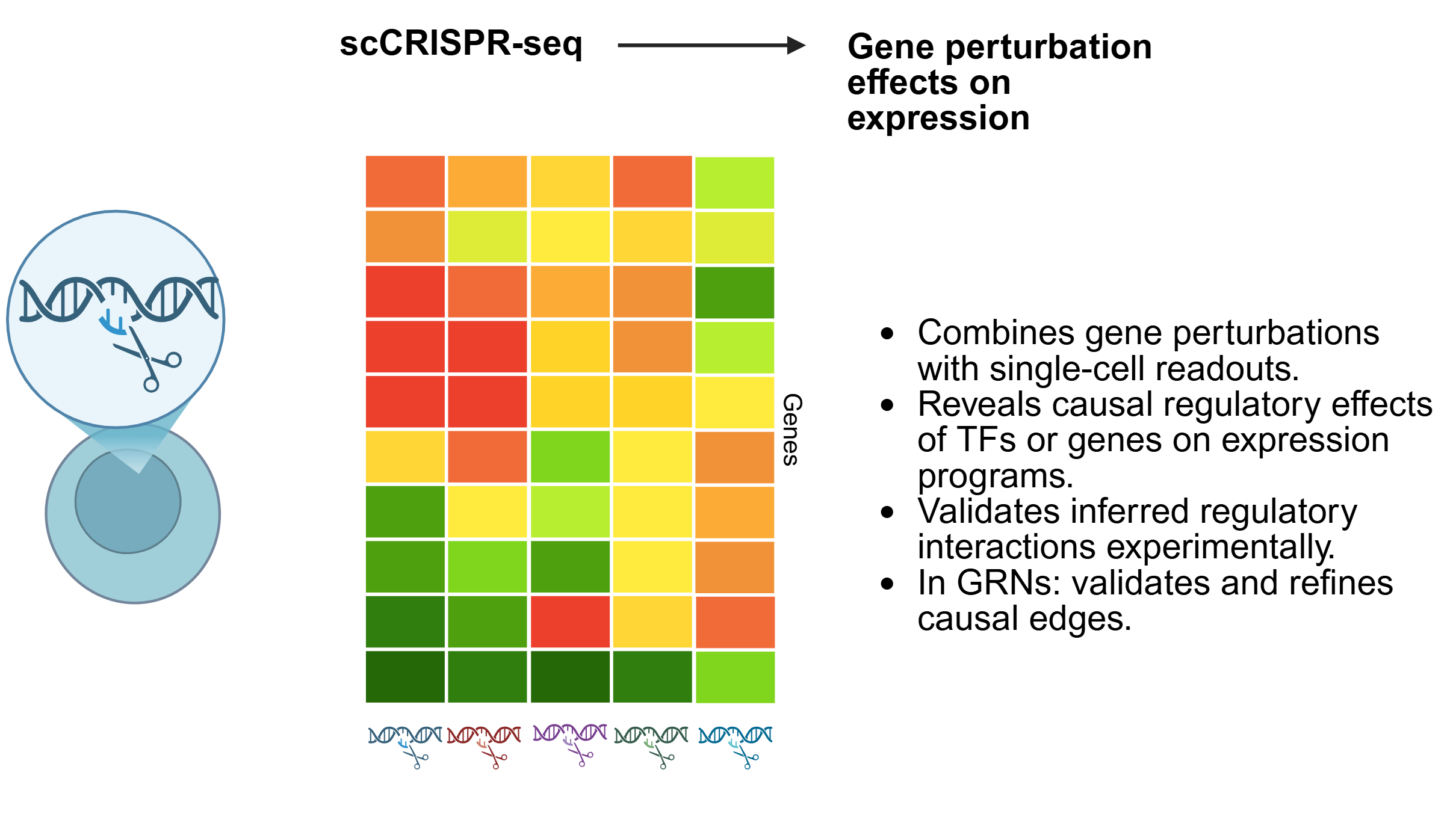
SC-MO-GRN-DB houses two primary types of data: reference networks and single-cell multiomics datasets. Reference networks capture transcription factor (TF) to target gene (TG) interactions, identified through TF localization via ChIP-seq with nearest-neighbor analysis, as well as TF perturbation studies such as knockouts. Single-cell multiomics datasets are often structured in a matrix format, where each column represents a single cell of a specific cell type.
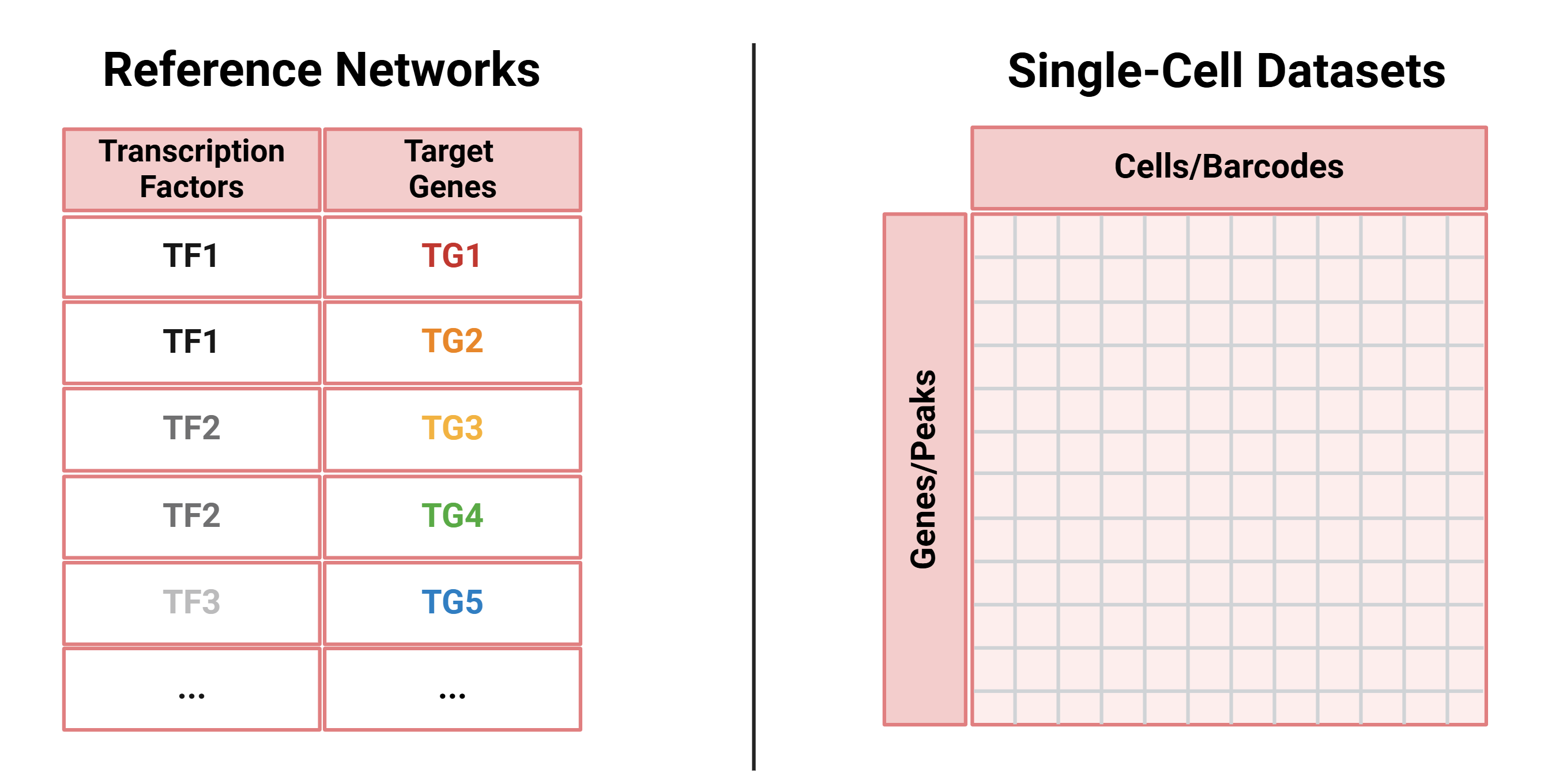
SC-MO-GRN-DB is designed to support flexible mapping between single-cell sequencing datasets and reference gene regulatory networks derived from independent sources. For a given tissue or cell type, the database may contain multiple single-cell datasets and multiple reference networks, reflecting different experimental conditions, modalities, and evidence types. These datasets are not intended to be paired in a one-to-one manner; instead, users may combine any compatible single-cell dataset with one or more reference networks when constructing or evaluating GRNs. We recommend leveraging all available datasets rather than relying on a single pairing, as this enables more robust and reproducible analyses. For benchmarking and method evaluation, an all-to-all strategy, testing each single-cell dataset against each reference network, allows users to assess consistency and performance across data sources. For biological discovery, results may be integrated across datasets and reference networks to construct consensus GRNs that capture shared regulatory signals.
For users interested in multi-modal GRN inference, SC-MO-GRN-DB provides guidance based on whether sequencing modalities are truly paired or unpaired. Jointly generated (paired) datasets bundle multiple modalities within a single dataset, allowing direct application of joint analysis tools such as CellOracle or LINGER without additional preprocessing. For unpaired datasets, each modality should be downloaded separately and integrated using established methods such as LIGER or Seurat CCA. Differences in experimental design and sequencing platforms can introduce batch effects and alignment challenges, so we recommend using truly paired datasets whenever possible. For all paired datasets in the database, barcode matching has already been performed, reducing the technical burden on users and facilitating downstream multi-modal GRN inference.